Epoxide Hydrolases
Expoxide-containing compounds are ubiquitously found in the environment from both natural and man-made sources, and a large variety of aromatic and alkenic compounds are also metabolized to epoxides endogenously. An epoxide (or oxirane) is a three membered cyclic ether which has specific reactivity patterns due to the highly polarized oxygen-carbon bonds in additional to a highly strained ring. Some reactive epoxides are responsible for electrophilic reactions with critical biological targets such as DNA and proteins, leading to mutagenic, toxic and carcinogenic effects. While most epoxides are of intermediate reactivity, are relatively stable at physiological pHs and do not present acute dangers to cells, they still need to be transformed in a controlled manner. The catalytic addition of water to epoxides or arene oxides by epoxide hydrolases (EH, E.C.3.3.2.3) to yield the corresponding 1,2-diols, or glycols, is only one of several ways that cells transform oxiranes. However, EHs are ubiquitous and hydration seems to be a common route of epoxide transformation. The reaction is energetically favorable with water as the only co-substrate.

The role of epoxide hydrolases seems to differ profoundly from organism to organism. Overall, these enzymes have three main functions: detoxification, catabolism and regulation of signaling molecules. In mammals, there are several epoxide hydrolases, including the soluble epoxide hydrolase (sEH) and the microsomal epoxide hydrolase (mEH).
Soluble epoxide hydrolase
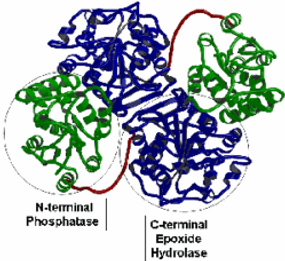
The soluble epoxide hydrolase (sEH; also known as the cytosolic epoxide hydrolase in older literature) is encoded by the EPHX2 gene that is localized on chromosome 8 in human. This 45 kb long gene comprises 19 exons that translate into a 555 aminoacid long protein. As shown on its crystal structure, the sEH protein is a homodimers, of ~62 kDa monomeric subunits with isoelectric points between 5 and 6. Each monomer is comprised of two distinct structural domains, linked by a proline-rich peptide segment (in red on the structure). The epoxide hydrolase activity resides in the ~35-kDa C-terminal domain (in blue), which contains an ?/?-hydrolase fold structure homologous to the bacterial haloalkane dehalogenase, the plant soluble EHs and the microsomal EH.
The roughly 25-kDa N-terminal domain (in green) contains a distinct ?/? fold topology belonging to the haloacid dehalogenase enzyme superfamily. The N-terminal domain catalytic site is a functional phosphatase. In addition, the N-terminal domain appears to serve a critical role in stabilization of the domain-swapped architecture of the dimer.
Like the other member of the ?/?-hydrolase fold family of proteins, the sEH is characterized by a nucleophile-histidine-acid catalytic triad and have a two-step mechanism involving the formation of a covalent intermediate. In the first step of the catalytic cycle of EHs, the epoxide quickly binds to the active site of the enzyme. The substrate epoxide is polarized by two tyrosine residues (382 and 465), which hydrogen bond with the epoxide oxygen.
At the same time, the nucleophilic carboxylic acid of Asp334, present on the side of the catalytic cavity opposite to the tyrosines, makes a backside attack on the epoxide, usually at the least sterically hindered and most reactive carbon. The nucleophilic acid is oriented and activated by His523, a second carboxylic amino acid (Asp495) and possibly other amino acids in the catalytic site for this attack.

The opening of the epoxide results in an ester bond between the enzyme carboxylic acid and one alcohol functionality of the diol.. This is termed the hydroxyl alkyl-enzyme intermediate (bottom right of Figure). Once the covalent hydroxyl alkyl-enzyme is formed, the histidine moves far enough from the nucleophilic acid (now ester) to allow a water molecule to be activated by the acid-histidine pair (bottom left of Figure). This very basic water attacks the carbonyl of the ester, releasing the diol product and the original enzyme.
Based on the catalytic mechanism we designed transition state mimics of the enzyme, optimized them to be low nano to picomolar level inhibitors, and used them to assist in structural work, map the internal domain of the enzyme, and develop good structure activity relationships. The SAR allowed us to predict that some environmental chemicals will be EH inhibitors including the industrial waste product dicyclohexyl urea, some herbicides (Diuron, Siduron) and the commonly used personal care product triclocarban.

Over the past decade, we have optimized the structures for potency and bioavailability/pharmacokinetics to evaluate the endogenous role of the sEH. In the last 5 years, we have increased the exposure/potency ratio of these compounds (area under the curve/concentration to inhibit 50% of the hydrolase activity, AUC/IC50) over 600x (see figure), and one of these inhibitors is in Phase I human clinical trials for hypertension by Arête Therapeutics. Multiple technologies had to be developed to carry out these studies, a totally new fluorogenic reporter was developed in order to have substrates of sufficient sensitivity to distinguish among inhibitors with IC50’s under 10nM. We also developed LC-MS/MS based analytical procedures where picomolar concentrations of the sEHIs could be monitored on 5 ?l of blood, allowing rapid ADME studies. These powerful inhibitors allowed us to determine endogenous roles for the sEH and may result in clinical drugs as well.
Over the past decade, using specific inhibitors developed in our laboratory as well as genetic knock-out animal, we found strong evidence for endogenous roles for the sEH. We demonstrated that the sEH is clearly implicated in the regulation of blood pressure, pain and inflammation through the hydrolysis of endogenous epoxide containing regulatory lipids in the arachidonic acid cascade such as epoxyeicosatrienoic acids (EETs).For example the over view of the arachidonate cascade (right) shows three major branches.

In the P450 branch one can make epoxides of arachidonic acid (EETs) which are anti-hypertensive, anti-inflammatory and analgesic. In contrast 20-HETE is a potent regulatory lipid increasing blood pressure and inflammation. Simplistically these two mediators work in opposition, however with age and disease the balance often shifts to 20-HETE. The major route of degradation of EETs is sEH catalyzed hydration to their corresponding diols (DHETs), but when sEH inhibitors (sEHI) block sEH, EETs are increased and blood pressure, inflammation and pain are reduced.
In a murine model of hypertension (564), we showed that treatment with a sEH inhibitor resulted in lower systolic blood pressure (at right). This was shown in various animal models of hypertension. The treatment with sEH inhibitor was also associated with the reduction of organ damages. As shown below, treatment with sEH inhibitor 950 or AUDA results in reduction and prevention of cardiac hypertrophy (630).



Microsomal epoxide hydrolase
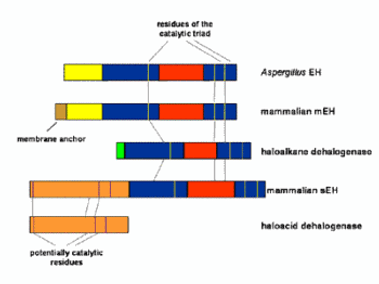
The microsomal epoxide hydrolase (mEH; also known as epoxide hydratase in older literature) is encoded by the EPHX1 gene that is localized on chromosome 1 in human. This gene translates into 455 amino acid residues corresponding to a ~50 kDa protein, with a strongly hydrophobic transmembrane anchor of approximately 20 residues at the N-terminal. The C-terminal domain, which contains the catalytic residues, is homologous to a haloalkane dehalogenase, like the soluble EH. As shown at right, the mEH is distantly related to the sEH, but the enzymes diverged at the level of prokaryotes. The mechanism of mEH is overall very similar to the mechanism described above for sEH. The structure of this enzyme has not yet be determined.
The mEH is well established in xenobiotic metabolism and its substrate selectivity generally complements that of the sEH. mEH inhibitors (mEHI) such as valpromide have resulted in adverse drug interactions. We are developing potent mEH inhibitors, which can be used for pharmacological studies to evaluate the in vivo role of the mEH in xenobiotic metabolism. We are using these inhibitors to test the hypothesis that there are roles for the enzyme in the metabolism of endogenous chemicals. Such inhibitors are needed to dissect the relative contribution of xenobiotic metabolizing enzymes, such as for the metabolism of naphthalene a common air pollutant.

Esterases and Amidases
Esterases and amidases catalyze the addition of a water molecule to an ester, thio-ester or an amide resulting in the formation of the corresponding acid and alcohol or amine. Like EHs and esterases are members of the ?/?-hydrolase fold family of enzymes. They share a common structure and catalytic mechanism involving the formation and hydrolysis of a covalent intermediate. Both enzymes add water to the substrate with no additional cofactors needed. While the EHs reaction is irreversible, the action of esterases and amidases is reversible and in some conditions these enzyme could be used for thesynthesis of ester or amides. Furthermore, the esterases and amidases used two substrates (ester and water) to form two products (acid and alcohol or amine).

Esterases and amidases play an important role in maintaining normal physiology and metabolism, detoxifying various drugs and environmental toxicants in living systems and are increasing important for chemical synthesis in industry.While we are focusing on the role of esterases and amidases in the metabolism of pyrethroid insecticides, we also investigate their role in the metabolism of endocannabinoids, and thus their role in inflammation and analgesia. As indicated in the table at right, we have cloned and super-expressed 15 human esterases and amidases. We are studying their role in the metabolism of numerous compounds, including pesticides, pharmaceutical drugs and endogenous compounds. To study these enzymes, we have developed an array of fluorescent substrates and a variety of chemical inhibitors.

Metabolism of pyrethroid insecticides
Pyrethroids are the dominant insecticides used nowdays. Although regarded as generally safe, human exposure can result in neuropathies. Long term effects have been suggested; multiple acute symptoms and even occasional deaths have been reported. The major pyrethroids (left) are esters, and many are sold as geometrically and/or optically rich isomers.

Cypermethrin for example has 8 optical and geometrical isomers with vastly different biological properties. We have a library of pyrethroids, have extensive expertise in their synthesis and analysis of these chiral esters using GLC-MS, LC-MS and biosensors, and we have prepared numerous immunoassaysbfor pyrethroids and metabolites. We developed a novel class of intensely fluorescent, stable, optically active surrogate substrates for pyrethroid esterase activity. They facilitate high throughput assays, monitoring biochemical purification and biomonitoring using very small samples.

The importance of esterases has been long recognized in toxicology. This is in part because carbamate and organophosphate insecticides could result in chemical knockouts. In the last few years the pyrethroid insecticides have emerged as the dominant insecticides and most are degraded largely by esterases. Thus, we are investigating the roles of esterases in their metabolism. Clearly genetic or chemical alterations in esterase could alter the risk not only of insecticides like pyrethroids but a variety of xenobiotics including common drugs and prodrugs as well as natural flavors and odors.
To test the hypothesis that hydrolysis of a single pyrethroid stereoisomer by carboxylesterases correlates to that of the corresponding
authentic pyrethroid stereoisomer, we synthesized all the stereoisomers of pyrethroid fluorescent substrates mimicking cypermethrin and fenvalerate. The hydrolysis of such stereoisomers by two purified murine hepatic carboxylesterases and human carboxylesterases (CES1 and CES2) are well correlated with hydrolysis of authentic pyrethroids in both activity and stereo-selectivity. Furthermore, using docking experiments, we explained the selectivity of CES1 for these compounds (figure below).

Metabolism of endocannabinoids
Endocannabinoids are substances produced from within the body which activate cannabinoid receptors (CB). The first such compound identified was arachidonoyl ethanolamide (also reported as anandamide). Anandamide is derived from the essential fatty acid arachidonic acid. It binds to the central (CB1) and, to a lesser extent, peripheral (CB2) cannabinoid receptors, where it acts as a partial agonist. It is found in nearly all tissues in a wide range of animals. Another endocannabinoid, 2-arachidonoyl glycerol, binds to both the CB1 and CB2 receptors with similar affinity, acting as a full agonist at both. 2-AG is present at significantly higher concentrations in the brain than anandamide, and thus it is though to be mostly responsible for endocannabinoid signalling in vivo. It binds primarily to the CB1 receptor, and only weakly to the CB2 receptor. More recently, N-arachidonoyl-dopamine (NADA) was found to preferentially bind to the CB1 receptor. Like anandamide, NADA is also an agonist for the vanilloid receptor subtype 1 (TRPV1), a member of the vanilloid receptor family.

one cell and activate the cannabinoid receptors present on other nearby cells. Although in this intercellular signaling role they are similar to the well-known monoamine neurotransmitters, such as acetylcholine, GABA or dopamine, endocannabinoids differ in numerous ways from them. For instance, they are have retrograde signaling. Furthermore, endocannabinoids are lipophilic molecules that are not very soluble in water. They are not stored in vesicles, and exist as integral constituents of the membrane bilayers that make up cells. They are believed to be synthesized 'on-demand' rather than made and stored for later use.
The main metabolism pathway of endocannabinoids is through hydrolysis by esterases and amidases, especially by FAAH and MGL. We are using our unique esterase inhibitors to ask the endogenous roles for these enzymes and other esterases in chemical mediation. Already we have shown (at right) that inhibition of 2-acylglycerol lipase (MGL) can stabilize a potent endocannabinoid and reduce the proliferation of human prostate cancer cells. Thus as shown at right, alterations in esterase activity can have profound biological effects through both xenobiotic and endogenous esters.
